
von Willebrand’s disease
อ.นพ.สันติ สิลัยรัตน์ คณะแพทยศาสตร์วชิรพยาบาล มหาวิทยาลัยนวมินทราธิราช
von Willebrand’s disease คือโรคทางกรรมพันธุ์ชนิดหนึ่งที่มีลักษณะทางคลินิกคือ การมีเลือดออกผิดปกติ โดยเกิดจากความผิดปกติของการเกาะยึด (adhesion) และก่อตัวรวมกัน (aggregation) ของเกล็ดเลือด โรคนี้ได้รับการบันทึกและบรรยายถึงลักษณะของโรคเอาไว้เป็นครั้งแรกโดย Erik von Willebrand ในปี ค.ศ. 1926 ซึ่งได้ตั้งข้อสังเกตไว้ว่าเป็นภาวะเลือดออกผิดปกติที่มีความแตกต่างจากโรคในกลุ่ม hemophilia และให้ชื่อโรคไว้ว่า hereditary pseudohemophilia หลังจากนั้นต่อมาอีกหลายปีจึงได้มีการค้นพบส่วนประกอบของเลือดที่สามารถแก้ไขความผิดปกติที่เกิดขึ้นได้ และให้ชื่อเรียกว่า von Willebrand factor (vWF)
von Willebrand factor เป็นโปรตีนที่จับกับคอลลาเจนในผนังหลอดเลือดเมื่อเกิดการบาดเจ็บของหลอดเลือด และเหนี่ยวนำให้เกิดกระบวนการเกาะติดและก่อตัวรวมกันของเกล็ดเลือด รวมถึงทำให้เกิดการเชื่อมโยงกับกระบวนการแข็งตัวของเลือดด้วยสารกลุ่ม coagulation factors ผ่านทาง factor VIII และกระบวนการอื่น ๆ เช่น กระบวนการอักเสบและการสร้างเส้นเลือดใหม่ (angiogenesis) ผู้ป่วยโรค von Willebrand’s disease จึงมีลักษณะสำคัญทางคลินิกคือ การมีเลือดออกง่ายในบริเวณของผิวหนังหรือเยื่อบุผิวต่าง ๆ หลังจากเกิดการบาดเจ็บของหลอดเลือด เช่น จากอุบัติเหตุ หรือการผ่าตัด การวินิจฉัยโรคนี้มักจะได้จากประวัติการมีเลือดออกผิดปกติของผู้ป่วยและคนในครอบครัวร่วมกับการตรวจวัดระดับหรือการทำงานของ vWF และสามารถให้การรักษาได้ด้วยยา desmopressin หรือการให้ vWF ในเลือดบริจาค เป็นต้น
ระบาดวิทยาและการจำแนกกลุ่มโรค
จากการศึกษาทางระบาดวิทยา ความชุกของ von Willebrand’s disease นั้นอยู่ระหว่าง 0.6-1.3% และพบในกลุ่มผู้ป่วยหญิงมากกว่าชาย โดยขึ้นอยู่กับระดับของ vWF ที่ใช้ในการวินิจฉัย (ค่าปกติในเลือดอยู่ระหว่าง 50-150 IU/dL) ส่วนใหญ่แล้วมักจะให้การวินิจฉัยโรคนี้หากมีระดับ vWF ในเลือดน้อยกว่า 30 IU/dL แต่ในแง่ของการตัดสินใจให้การรักษามักจะพิจารณาเมื่อมีระดับในเลือดน้อยกว่า 40 IU/dL
von Willebrand’s disease นั้นแบ่งย่อยออกได้เป็นหลายกลุ่มคือ type 1, 2 และ 3 โดยในแต่ละกลุ่มมีลักษณะความผิดปกติที่สำคัญคือ
- Type 1 von Willebrand’s disease (พบได้ราว 70-80% ของผู้ป่วยทั้งหมด) เป็นกลุ่มที่มีความผิดปกติหลักคือ การมีปริมาณ vWF ลดลง (quantitative deficiency)
- Type 2 von Willebrand’s disease (พบได้ราว 20%) เป็นกลุ่มที่มีความผิดปกติในแง่การทำงานของ von Willebrand factor เป็นหลัก และอาจมีปริมาณของ vWF ในเลือดปกติหรือลดลงร่วมด้วยก็ได้
- Type 3 von Willebrand’s disease เป็นกลุ่มที่มีความรุนแรงของโรคมากที่สุด โดยเป็นกลุ่มที่ไม่มีการสร้าง vWF ในเลือดเลย พบได้น้อยกว่า 5% ของผู้ป่วยทั้งหมด
ลักษณะทางคลินิก
ลักษณะและความรุนแรงของอาการในผู้ป่วย von Willebrand’s disease นั้นมีความแตกต่างกันไปในผู้ป่วยแต่ละรายขึ้นอยู่กับชนิดของโรค ระดับของ vWF ในเลือด และปัจจัยอื่น ๆ เช่น เพศและอายุของผู้ป่วย สำหรับในผู้ป่วยเด็ก อาการที่มักจะนำผู้ป่วยมาพบแพทย์ ได้แก่ อาการจ้ำเลือดออกตามตัวและเลือดกำเดาไหล (epistaxis) สำหรับในผู้ใหญ่มักเป็นอาการเลือดออกสะสมตามจุดต่าง ๆ (hematoma), เลือดประจำเดือนออกมากผิดปกติ (menorrhagia) และเลือดออกมากผิดปกติจากบาดแผลต่าง ๆ โดยส่วนใหญ่ (ราว 60-80%) มักจะพบเมื่อรับการผ่าตัดหรือถอนฟัน สำหรับในผู้สูงอายุที่เป็นโรค von Willebrand’s disease type 2 หรือ 3 อาจมีอาการเลือดออกในทางเดินอาหารจากรอยโรค angiodysplasia ได้ เป็นต้น ส่วนอาการเลือดออกผิดปกติอื่น ๆ ที่พบได้ ได้แก่ อาการเลือดออกในข้อ (intra-articular bleeding) ซึ่งพบได้ในผู้ป่วยกลุ่ม type 3 เนื่องจากกลุ่มนี้มักจะมีระดับหรือความผิดปกติของ factor VIII ร่วมด้วย ผู้ป่วยที่มีเลือดออกในข้อมักจะเกิดความผิดปกติของข้อในระยะยาวทำให้เกิดอาการเจ็บปวดและคุณภาพชีวิตที่แย่กว่าผู้ป่วยกลุ่มอื่น ๆ
สำหรับในผู้ป่วยหญิง อาการสำคัญที่พบได้แก่ อาการเลือดประจำเดือนออกมากผิดปกติ อาการตกเลือดหลังคลอดทั้งชนิดปฐมภูมิและทุติยภูมิ (primary and secondary postpartum hemorrhage) ซึ่งในบางรายอาจทำให้เกิดภาวะโลหิตจาง หรือมีอาการรุนแรงจนอาจต้องได้รับการรักษาด้วยการตัดมดลูกได้ นอกจากนี้ในผู้ป่วยบางรายยังอาจมาด้วยอาการปวดท้องน้อยเนื่องจากมีเลือดออก ovarian cysts ได้
โดยทั่วไปแล้วเมื่อผู้ป่วยโรค type 1 von Willebrand’s disease มีอายุมากขึ้นจะมีระดับ vWF ในเลือดเพิ่มขึ้นจนในบางรายอาจมีระดับเท่ากับคนปกติได้ แต่จากการศึกษาเปรียบเทียบลักษณะอาการเลือดออกผิดปกติที่เกิดขึ้นในผู้ป่วยเหล่านี้เมื่ออายุเกิน 65 ปีขึ้นไปเทียบกับกลุ่มที่มีอายุน้อยกว่า กลับพบว่าไม่ได้มีความแตกต่างกันอย่างมีนัยสำคัญ ซึ่งแสดงให้เห็นว่าการมีระดับ vWF มากขึ้นไม่ได้ช่วยทำให้โอกาสในการเกิดภาวะเลือดออกผิดปกติลดลง ข้อสังเกตที่น่าสนใจประการหนึ่งสำหรับผู้ป่วยกลุ่มนี้ซึ่งพบในการศึกษาไม่นานมานี้ก็คือ เมื่อผู้ป่วยกลุ่มนี้อายุมากขึ้น ความเสี่ยงของการเกิดโรคหัวใจและหลอดเลือดกลับน้อยกว่าคนปกติในกลุ่มอายุเดียวกัน
ลักษณะทางพยาธิสรีรวิทยาของโรค
vWF mutations ในผู้ป่วย von Willebrand’s disease ความผิดปกติของยีนที่ควบคุมการสร้าง von Willebrand factor (vWF gene) เกิดจากกระบวนการ mutation ของยีน ซึ่งมีผลทำให้มีการสร้าง vWF ในเชิงคุณภาพหรือปริมาณที่ลดลง หรือไม่มีการสร้างเลย และสามารถถ่ายทอดทางพันธุกรรมในครอบครัวเป็นได้ทั้งแบบ autosomal dominant และ autosomal recessive pattern โดยผู้ป่วยส่วนใหญ่มีการถ่ายทอดแบบ autosomal dominant pattern สำหรับในผู้ป่วย type 3 von Willebrand’s disease นั้นส่วนใหญ่ vWF gene มีการขาดหายไปทั้งหมด (null alleles) ซึ่งมีผลทำให้ไม่มีการสร้าง von Willebrand factor ขึ้นเลย นอกจากนี้การเกิด vWF mutation ยังอาจแสดงออกมาในรูปแบบของความผิดปกติในกระบวนการอื่น ๆ ทางสรีรวิทยาได้ด้วยเช่น ทำให้กระบวนการ intracellular routing, storage และ secretion ของ von Willebrand factor ผิดปกติ หรือทำให้มีกระบวนการสลาย vWF เร็วขึ้นกว่าปกติได้
Other genetic modifiers ผู้ป่วยโรค von Willebrand’s disease ส่วนหนึ่งมีอาการเลือดออกผิดปกติและมีระดับ vWF ลดลง แต่ตรวจไม่พบความผิดปกติของ vWF gene ซึ่งบ่งชี้ว่าอาจมีความผิดปกติของสารพันธุกรรมอื่น ๆ ที่ทำให้เกิดลักษณะความผิดปกติแบบเดียวกันนี้ได้ ตัวอย่างความผิดปกติที่เกี่ยวข้องที่พบ ได้แก่ หมู่เลือด ABO group ซึ่งพบว่าคนที่มีเลือดหมู่ O จะมีระดับ vWF ในเลือดน้อยกว่าคนที่มีเลือดหมู่อื่น ๆ ประมาณ 25% และยีนที่ควบคุมการสร้างโปรตีนในกระบวนการขับหรือกำจัด vWF เช่น C-type lectin domain family 4, member M (CLEC4M) หรือ syntaxin-binding protein 5 (STXBP5) เป็นต้น
การวินิจฉัย
โดยทั่วไปแล้วการวินิจฉัย von Willebrand’s disease สามารถทำได้โดยอาศัยประวัติอาการเลือดออกผิดปกติทั้งในผู้ป่วยและคนในครอบครัวร่วมกับการตรวจทางห้องปฏิบัติการพบความผิดปกติของ vWF หรือ factor VIII หรือทั้ง 2 อย่าง การค้นหาและประเมินลักษณะของอาการเลือดออกผิดปกติ ทั้งในแง่ของตำแหน่ง ความถี่ และความรุนแรงจากประวัติของผู้ป่วยเองหรือประวัติบุคคลอื่น ๆ ในครอบครัว สามารถทำได้โดยใช้แบบประเมินลักษณะของความผิดปกติ ทั้งในรูปแบบสอบถาม และแบบการให้คะแนน (scoring system) ซึ่งในทางปฏิบัติมีอยู่หลายแบบ เช่น แบบประเมินสร้างขึ้นและนำมาใช้โดย International Society on Thrombosis and Haemostasis (ISTH) (https://bh.rockefeller.edu/ISTH-BATR/) เป็นต้น แบบประเมินแต่ละชนิดมีข้อดีและข้อจำกัดอยู่บ้าง เช่น การใช้ประวัติเลือดออกผิดปกติหลังการทำหัตถการทางทันตกรรมหรือการผ่าตัด อาจพบได้น้อยในผู้ป่วยเด็กเล็กเนื่องจากยังไม่ถึงช่วงอายุที่มักจะมีการทำหัตถการต่าง ๆ ดังกล่าว เป็นต้น
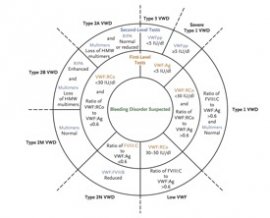
รูปที่ 1 แผนภูมิการตรวจต่าง ๆ เพื่อการวินิจฉัยโรค von Willebrand’s disease
(ภาพจาก Leebeek F WG, Eikenboom J CJ. N Engl J Med 2016;375:2067-80.)
ในแง่ของการตรวจทางห้องปฏิบัติการเพื่อการวินิจฉัยโรค von Willebrand’s disease นั้นมีการตรวจอยู่ 3 ชนิด ได้แก่ การตรวจ vWF antigen; การตรวจ vWF-dependent platelet adhesion (ด้วยวิธี von Willebrand factor-ristocetin cofactor activity [vWF-RCo] assay) และการตรวจ factor VIII activity ในกรณีที่ตรวจวัดระดับ von Willebrand factor antigen พบว่าระดับต่ำกว่า 5 IU/dL จะให้การวินิจฉัยว่าเป็น type 3 von Willebrand's disease อย่างไรก็ตาม มีผู้ป่วย type 1 von Willebrand’s disease บางรายที่อาจมีระดับ vWF antigen น้อยกว่า 5 IU/dL ได้ ดังนั้น ในการตรวจแยกโรคอาจทำได้โดยการตรวจวัดระดับ vWF propeptide ซึ่งเป็นโปรตีนที่เกิดขึ้นในระหว่างการสร้าง vWF เพิ่มได้ หากพบว่าโปรตีนชนิดนี้ต่ำหรือไม่มีเลยก็จะวินิจฉัยว่าเป็น type 3 ส่วนในรายที่ยังพบว่าปกติหรือลดลงเล็กน้อยก็ให้การวินิจฉัยเป็น type 1 von Willebrand's disease
ผู้ป่วยที่มี vWF antigen ในระดับที่ตรวจวัดได้ ควรทำการตรวจ vWF-RCo assay เพิ่มเติม ซึ่งการตรวจนี้เป็นการตรวจในแง่ของการทำงานของ vWF โดยการจับกับเกล็ดเลือด หากระดับ vWF antigen ที่ลดลงสัมพันธ์กันกับความสามารถในการจับกับเกล็ดเลือดที่ลดลงก็จะให้การวินิจฉัยว่าเป็น type 1 von Willebrand’s disease แต่หากความสามารถในการจับกับเกล็ดเลือดลดลงไม่สัมพันธ์กับระดับของ vWF antigen ที่ลดลง กล่าวคือ vWF-RCo activity to vWF antigen ratio ≤ 0.6 ก็จะให้การวินิจฉัยว่าเป็น type 2 von Willebrand’s disease
หลังจากที่สามารถให้การวินิจฉัย type 2 von Willebrand’s disease แล้ว สามารถทำการตรวจเพื่อแยกชนิดย่อยคือ type 2A, 2B และ 2M von Willebrand’s disease ได้ด้วยการตรวจเพิ่มเติมต่าง ๆ ได้แก่ การตรวจวัด von Willebrand factor multimers และ ristocetin-induced platelet aggregation ส่วน type 2N von Willebrand’s disease นั้นสามารถทำได้โดยการตรวจ factor VIII activity ซึ่งหากพบว่า factor VIII activity to vWF antigen ratio ≤ 0.6 ก็จะวินิจฉัยว่าเป็น type 2N von Willebrand’s disease ได้
สำหรับการตรวจ vWF gene นั้น ในปัจจุบันยังไม่ได้มีการนำมาใช้เพื่อการวินิจฉัยในทางเวชปฏิบัติทั่วไป แต่อาจจะมีประโยชน์ในแง่ของการแยกชนิดของ type 2N และ 2B disease รวมถึงการวินิจฉัย type 3 disease ซึ่งสามารถนำไปเป็นข้อมูลประกอบในการให้คำปรึกษาและวางแผนครอบครัวได้
การรักษา
หลักการในการรักษาโรค von Willebrand’s disease ได้แก่ การทำให้ระดับ vWF และ factor VIII ให้กลับเป็นปกติในระหว่างที่เกิดภาวะเลือดออกหรือก่อนที่จะมีการทำหัตถการต่าง ๆ ซึ่งสามารถทำได้โดยการให้ยา desmopressin หรือโดยการให้ vWF concentrate หรือ factor VIII-vWF concentrate ที่ผลิตแยกออกมาเฉพาะ ทั้งนี้ขึ้นกับชนิดของโรคที่พบในผู้ป่วย ดังตารางที่ 1
ตารางที่ 1 การรักษาสำหรับโรค von Willebrand’s disease ชนิดต่าง ๆ
Desmopressin เป็นการรักษาสำหรับผู้ป่วย type 1 และ type 2 von Willebrand’s disease หรือในผู้ป่วยที่ตรวจพบว่ามีระดับ vWF ต่ำกว่าปกติ ยานี้สามารถให้ได้ทั้งในรูปแบบฉีดเข้าทางหลอดเลือดดำ พ่นทางจมูก และฉีดเข้าใต้ผิวหนัง สามารถทำให้ระดับ vWF และ factor VIII ได้ประมาณ 2-4 เท่า และสามารถให้ซ้ำได้ทุก 12-24 ชั่วโมง โดยพิจารณาตามการตอบสนองต่อการรักษาของผู้ป่วยแต่ละราย อาการข้างเคียงจากยา desmopressin โดยทั่วไปค่อนข้างน้อยและไม่รุนแรง ได้แก่ ความดันโลหิตลดต่ำ และ hyponatremia ซึ่งในกรณีของ hyponatremia สามารถป้องกันได้โดยให้ผู้ป่วยจำกัดปริมาณน้ำดื่มให้เหลือน้อยกว่า 1,500 mL ต่อวันก่อนให้ยา
Factor concentrate เป็นการรักษาที่เหมาะสมสำหรับผู้ป่วยในกลุ่ม type 2 และ type 3 von Willebrand’s disease เนื่องจาก factor concentrate ที่ผลิตขึ้นได้มักจะมีความเข้มข้นของปัจจัยการแข็งตัวของเลือดที่แตกต่างกันไป ดังนั้น ขนาดที่ใช้ในการรักษาผู้ป่วยจึงขึ้นกับความเข้มข้นของปัจจัยการแข็งตัวของเลือดนั้น ๆ และขึ้นกับระดับ vWF หรือระดับการทำงานของ vWF-RCo activity เป้าหมายที่ต้องการ และพิจารณาจากความรุนแรงของอาการเลือดออกหรือหัตถการที่วางแผนจะทำในผู้ป่วย ดังตารางที่ 2
ตารางที่ 2 ขนาดของการใช้ factor concentrate สำหรับผู้ป่วย von Willebrand’s disease
โดยทั่วไปแล้วเมื่อให้ vWF ชนิด high purity concentrate โดยไม่มี factor VIII มักจะสามารถทำให้ระดับ factor VIII และ factor VIII activity เพิ่มขึ้นได้ด้วยเนื่องจากเมื่อมี vWF ในร่างกายเพิ่มมากขึ้นจะเป็นตัวกระตุ้นให้มีการสร้าง factor VIII มาจับกับ vWF มากขึ้นด้วย ดังนั้น ในกรณีที่ไม่ใช่ภาวะเลือดออกเฉียบพลันสามารถให้เฉพาะ vWF concentrate อย่างเดียวได้ แต่หากต้องให้ห้ามเลือดในทันทีอาจพิจารณาให้ vWF และ factor VIII ไปพร้อมกันได้ เพื่อให้สามารถออกฤทธิ์ห้ามเลือดได้เร็วขึ้น
ในกรณีที่ผู้ป่วยต้องเข้ารับการผ่าตัดใหญ่ และการคลอดบุตรซึ่งมักจะมีการเสียเลือดได้ค่อนข้างมาก ควรตั้งเป้าหมายให้มี vWF-RCo และ factor VIII activity มากกว่า 100 IU/dL ขึ้นไป เพื่อให้มั่นใจได้ว่าจะไม่เกิดภาวะเลือดออกมากผิดปกติ โดยอาจให้ทั้ง factor concentrate และ desmopressin ร่วมกันก็ได้ และหลังจากที่เสร็จสิ้นการผ่าตัดแล้วควรมีการตรวจติดตามระดับ vWF-RCo และ factor VIII activity ต่อเป็นระยะ ๆ โดยตั้งเป้าหมายให้มากกว่า 50 IU/dL ต่อไปอีก 7-10 วัน
ในปัจจุบันมีการคิดค้น vWF ชนิดสังเคราะห์หรือ recombinant vWF concentrate ขึ้นมาใช้แล้ว ซึ่งพบว่าสามารถออกฤทธิ์ห้ามเลือดได้ดีเท่ากันกับ vWF ที่ได้จากเลือด และมีข้อดีคือ มีความเสี่ยงต่อการติดเชื้อจากเลือดบริจาคที่น้อยกว่าและมีโอกาสเกิดอาการแพ้น้อย ในปี ค.ศ. 2015 ที่ผ่านมา recombinant vWF concentrate ได้รับอนุมัติจาก US FDA ให้สามารถนำมาใช้ในการรักษาภาวะเลือดออกมากจากโรค von Willebrand’s disease ในผู้ใหญ่แล้ว
การรักษาในเชิงป้องกัน (prophylactic treatment)
สำหรับในกรณีของ von Willebrand’s disease โดยทั่วไปยังไม่ได้มีการแนะนำให้รักษาผู้ป่วยโรคนี้หากยังไม่เกิดอาการเลือดออกผิดปกติ อย่างไรก็ตาม มีการศึกษาเมื่อไม่นานมานี้แบบ prospective dose-escalating study เพื่อศึกษาเกี่ยวกับบทบาทของการรักษาในแบบ prophylactic treatment พบว่าผู้ป่วยที่ได้รับการรักษาโดยให้มี vWF-RCo activity 50 IU/kg สัปดาห์ละ 2-3 ครั้ง สามารถทำให้จำนวนครั้งของอาการเลือดออกต่าง ๆ เช่น เลือดออกในทางเดินอาหาร ในข้อ เลือดกำเดาไหลรุนแรงลดลงได้อย่างชัดเจน แต่เนื่องจากการศึกษานี้เป็นการศึกษาที่ยังมีผู้เข้าร่วมการศึกษาน้อย (11 คน) จึงยังไม่สามารถบอกถึงประสิทธิภาพของการรักษาด้วยวิธีนี้ได้ และจำเป็นต้องรอผลการศึกษาเพิ่มเติมต่อไป
การรักษาสำหรับผู้ป่วยทางนรีเวช
สำหรับผู้ป่วยโรค von Willebrand’s disease หญิงที่มีปัญหาเลือดประจำเดือนออกมากควรเริ่มต้นให้การรักษาด้วยยาคุมกำเนิดที่มีทั้งฮอร์โมน progestin และ estrogen ก่อน จากนั้นอาจพิจารณาให้ยา tranexamic acid เพิ่มเติมเพื่อช่วยลดอาการเลือดออก สำหรับในรายที่เลือดออกมากอาจพิจารณาการใช้ยา desmopressin และ/หรือ factor concentrates เพิ่มเติมได้ในรายที่ไม่ตอบสนองต่อการรักษาข้างต้น การพิจารณาให้การรักษาอื่น เช่น endometrial ablation หรือ hysterectomy ได้
สำหรับผู้ป่วยหญิงที่ตั้งครรภ์ กลุ่ม type 1 von Willebrand’s disease นั้นมักจะมีระดับ vWF ในเลือดที่เพิ่มขึ้นในช่วงใกล้คลอดซึ่งทำให้โอกาสในการเกิดเลือดออกผิดปกติลดลงในระยะคลอด ยกเว้นในรายที่มีระดับ vWF ต่ำมากอาจจะต้องให้การรักษาด้วยยา desmopressin หลังจากที่คลอดแล้ว แต่สำหรับกลุ่ม type 2 จะยังมีการทำงานของ vWF ที่ต่ำอยู่แม้จะมีระดับ vWF เพิ่มขึ้น ดังนั้น ผู้ป่วยกลุ่มนี้จึงอาจต้องได้รับการรักษาเพิ่มเติมเพื่อป้องกันการตกเลือดหลังคลอด โดยการให้ factor concentrates นั้นมีวิธีการให้แบบเดียวกันกับในการรักษาผู้ป่วยที่ต้องเข้ารับการผ่าตัดใหญ่
References
- von Willebrand E. Hereditar pseudo-hemofili. Fin Lakaresallsk Handl 1926;68:87-112.
- Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood 2014;124:1412-25.
- Zimmerman TS, Ratnoff OD, Powell AE. Immunologic differentiation of classic hemophilia (factor 8 deficiency) and von Willebrand’s disease, with observations on combined deficiencies of antihemophilic factor and proaccelerin (factor V) and on an acquired circulating anticoagulant against antihemophilic factor. J Clin Invest 1971;50:244-54.
- Lenting PJ, Casari C, Christophe OD, Denis CV. von Willebrand factor: the old, the new and the unknown. J Thromb Haemost 2012;10:2428-37.
- Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (vWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia 2008;14:171-232.
- Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood 1987;69:454-9.
- Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand’s disease. Thromb Haemost 2000;84:160-74.
- Laffan MA, Lester W, O’Donnell JS, et al. The diagnosis and management of von Willebrand's disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol 2014;167:453-65.
- Castaman G, Goodeve A, Eikenboom J. Principles of care for the diagnosis and treatment of von Willebrand's disease. Haematologica 2013;98:667-74.
- Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand's disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost 2006;4:2103-14.
- Cuker A, Connors JM, Katz JT, Levy BD, Loscalzo J. A bloody mystery. N Engl J Med 2009;361:1887-94.
- Sanders YV, Fijnvandraat K, Boender J, et al. Bleeding spectrum in children with moderate or severe von Willebrand's disease: relevance of pediatric-specific bleeding. Am J Hematol 2015;90:1142-8.
- de Wee EM, Sanders YV, Mauser-Bun-schoten EP, et al. Determinants of bleeding phenotype in adult patients with moderate or severe von Willebrand's disease. Thromb Haemost 2012;108:683-92.
- Silwer J. von Willebrand’s disease in Sweden. Acta Paediatr Scand Suppl 1973;238:1-159.